Caution
These documents refer to an obsolete way of installing and running FALCON. They will remain up for historical context and for individuals still using the older version of FALCON/FALCON_unzip.
Attention
The current PacBio Assembly suite documentation which includes new bioconda instructions for installing FALCON, FALCON_unzip and their associated dependencies can be found here pb_assembly

About FALCON¶
Overview¶
FALCON and FALCON-Unzip are de novo genome assemblers for PacBio long reads, also known as
single-molecule real-time (SMRT) sequences. FALCON is a diploid-aware assembler
which follows the hierarchical genome assembly process (HGAP) and is optimized for
large genome assembly (e.g. non-microbial). FALCON produces a set of primary contigs (a-contigs),
which represent divergent allelic variants. Each a-contig is associated with a homologous
genomic region on an p-contig.
FALCON-Unzip is a true diploid assembler. It takes the contigs from
FALCON and phases the reads based on heterozygous SNPs identified in the initial
assembly. It then produces a set of partially phased primary contigs and fully phased
haplotigs which represent divergent haplotyes.
Detailed Description¶
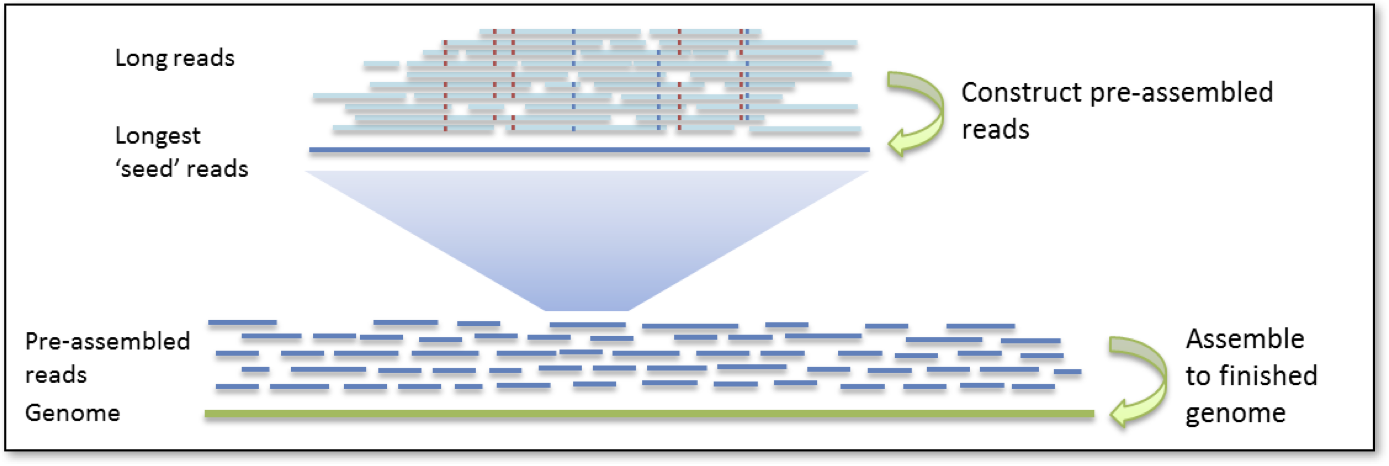
The hierarchical genome assembly process proceeds in two rounds. The first round of assembly involves the selection of seed reads, or the longest reads in the dataset (user-defined length_cutoff). All shorter reads are aligned to the seed reads, in order to generate consensus sequences with high accuracy. We refer to these as pre-assembled reads but they can also be thought of as “error corrected” reads. During the pre-assembly process, seed reads may be split or trimmed at regions of low read coverage (user-defined min_cov for falcon_sense_option). The performance of the pre-assembly process is captured in the pre-assembly stats file.
In the next round of HGAP, the preads, are aligned to each other and assembled into genomic contigs.
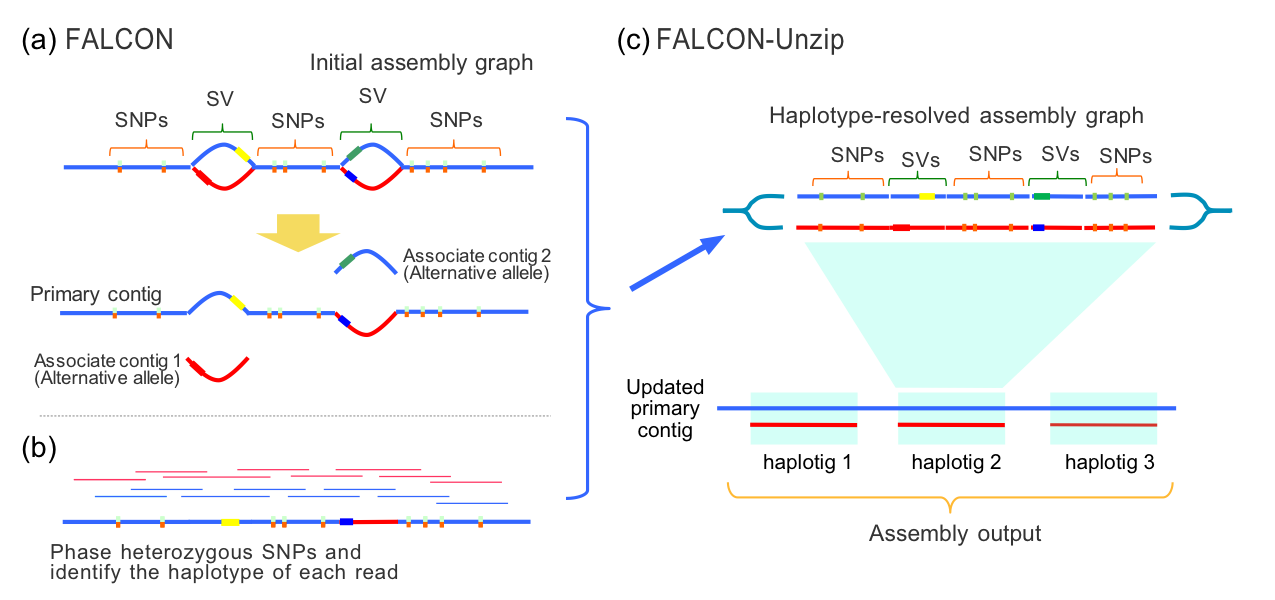
For more complex genomes assembled with FALCON,
“bubbles” in the contig-assembly graph that result from structural variation between haplotypes may be resolved as associate
and primary contigs. The unzip process will extend haplotype phasing beyond “bubble” regions, increasing the amount of phased
contig sequence. It is important to note that
while individual haplotype blocks are phased, phasing does not extend between haplotigs. Thus, in part C) of the
figure above, haplotig_1 and haplotig_2 may originate from different parental haplotypes. Additional information is
needed to phase the haplotype blocks with each other.
Associate contig IDs contain the name of their primary contig but the precise location of alignment must be determined with third party
tools such as NUCmer. For example, in a FALCON assembly, 000123F-010-01 is an associated contig to primary contig
000123F. In a FALCON-Unzip assembly, 000123F_001 is a haplotig of primary contig 000123F.
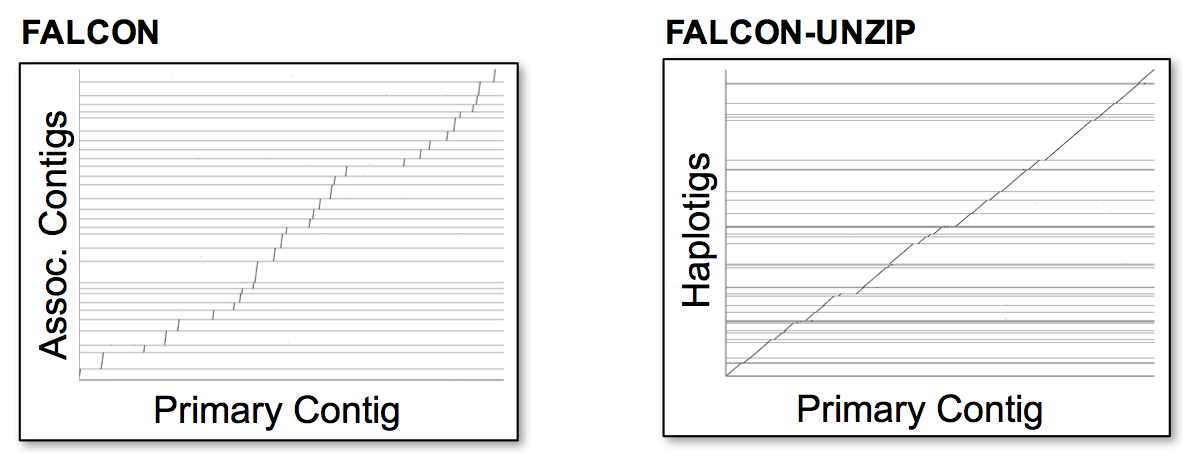
Below are examples of alignments between associate and primary contigs from FALCON, and haplotigs and primary contigs
from FALCON-Unzip. Alignments were built with NUCmer and visualized with Assemblytics. Precise coordinates
may be obtained with the show-coords utilty from MUMmer.
Choosing an Assembler: HGAP4 vs FALCON vs FALCON-Unzip¶
HGAP4¶
We recommend HGAP4, part of the SMRT Link web-based analysis suite, for genomes of known complexity, no larger than
human (3Gb or
smaller),
although underlying
compute resources for your SMRT Link instance will influence performance and feasibility. The assembly
process for HGAP4 in the SMRT Link GUI (graphical user interface) is identical to FALCON at the command line, besides
differences in
compute resource configuration and minor differences in directory structure. The HGAP4 pipeline by default includes a round of
genome “polishing”
which employs the resequencing pipeline.
HGAP4 RESULTS ARE NOT COMPATIBLE WITH FALCON-Unzip AT THIS TIME!
HGAP4 inputs are a PacBio subread BAM dataset, either Sequel or RSII. The FASTA and FASTQ files output from HGAP4 are a concatenation of the primary
and associate contigs, which are output from FALCON as separate files.
Command Line¶
Users more comfortable at the command line may use FALCON for genomes of any size
or complexity. Command line inputs are FASTA files of Sequel or RSII subreads. Command-line FALCON does not automatically polish the assembly. If a user
wishes, assembly polishing may
be run using the resequencing pipeline of pbsmrtpipe (available for command-line installation using the SMRT_Link download, see
SMRT_Tools_Reference_Guide for
installation instructions). Resequencing requires PacBio subread BAM inputs.
We recommend the FALCON-Unzip module for heterozygous or outbred organisms that are diploid or higher ploidy. Users wishing to run
FALCON-Unzip must do so only after running FALCON on the
command line. HGAP4 IS NOT COMPATIBLE WITH FALCON-UNZIP! The FALCON-Unzip module requires both FASTA and PacBio BAM inputs for subreads.